
COVID-19 caused by SARS-CoV-2 has become a global pandemic requiring the development of interventions for the prevention or treatment to curtail mortality and morbidity. No vaccine to boost mucosal immunity, or as a therapeutic, has yet been developed to SARS-CoV-2. In this study, we discover and characterize a cross-reactive human IgA monoclonal antibody, MAb362. MAb362 binds to both SARS-CoV and SARS-CoV-2 spike proteins and competitively blocks ACE2 receptor binding, by overlapping the ACE2 structural binding epitope. Furthermore, MAb362 IgA neutralizes both pseudotyped SARS-CoV and SARS-CoV-2 in 293 cells expressing ACE2. When converted to secretory IgA, MAb326 also neutralizes authentic SARS-CoV-2 virus while the IgG isotype shows no neutralization. Our results suggest that SARS-CoV-2 specific IgA antibodies, such as MAb362, may provide effective immunity against SARS-CoV-2 by inducing mucosal immunity within the respiratory system, a potentially critical feature of an effective vaccine.
Download PDF
Introduction
In December 2019, a novel coronavirus (SARS-CoV-2) was identified as the cause of an outbreak of acute respiratory infections. The coronavirus disease 2019 (COVID-19) ranges from mild to severe acute respiratory infection, with a fatality rate estimated to range from 2 to 3%1,2,3,4. Within 3 months of the first report cases, COVID-19 rapidly disseminated through the human population and had become a global pandemic by March 2020. Phylogenic analysis has classified SARS-CoV-2 within the sarbecoviruses subgenus, the β lineage that also contains SARS-CoV, sharing ~79.6% sequence identity4.
Interventions for the prevention or treatment of COVID-19 are crucial for the ongoing outbreak. Pre- or post-exposure immunotherapies with neutralizing antibodies, would be of great use by providing immediate mucosal immunity against SARS-CoV-2. Although concerns, as occurred with SARS-CoV5,6, that vaccines may cause disease enhancement still need to be addressed. The feasibility of human monoclonal antibodies (MAbs) as immunoprophylaxis or therapy against coronaviruses including SARS-CoV7,8,9,10 and MERS-CoV11 has been demonstrated. These anti-coronavirus MAbs primarily target the viral spike (S) glycoprotein, a type I transmembrane glycoprotein that produces recognizable crown-like spike structures on the virus surface. The receptor-binding domain (RBD) of the S protein facilitates viral entry into human cells through human angiotensin-converting enzyme 2 (ACE2) receptor binding leveraging a similar mechanism as SARS-CoV12,13,14.
Most current anti-SARS-CoV MAbs neutralize virus by binding to epitopes on the spike protein RBD of SARS-CoV15. We and others have demonstrated that neutralizing MAbs that block RBD-ACE2 binding could confer potent protection against SARS-CoV as both prophylaxis and treatment in various animal models7,9,10. Several anti-SARS-CoV MAbs have demonstrated cross-neutralizing activities against the S protein of SARS-CoV-216,17.
Antibody-dependent enhancement of viral infections are one of the major hurdles in the development of effective vaccines. This enhancement is likely facilitated by the Fc domain of IgG but not for its isotype variant IgA18. The avidity of mucosal IgA, in comparison with IgG, owing to the multimeric structure, enhances the antibody binding with antigens. In addition, the diverse, high level of glycosylation of IgA antibodies, further protects the mucosal surface with non-specific interference. In animal models, high titers of mucosal IgA in the lung is correlated with reduced pathology upon viral challenge with SARS-CoV19. How precisely which isotype may protect the mucosa from SARS-CoV-2 infection remains an open question.
In the current study, we describe the discovery of a cross-neutralizing human IgA monoclonal antibody, MAb362 IgA. This IgA antibody binds to SARS-CoV-2 RBD with high affinity competing at the ACE2 binding interface by blocking interactions with the receptor. MAb362 IgA neutralizes both pseudotyped SARS-CoV and SARS-CoV-2 in 293 cells expressing ACE2. The secretory IgA form of MAb326 also neutralizes authentic SARS-CoV-2 virus. Our results demonstrate that the IgA isotype may play a critical role in SARS-CoV-2 neutralization.
Results
Selection of MAb binding to RBD of SARS-CoV-2 in ELISA
We have previously developed and characterized a panel of human MAbs that targets the RBD of the SARS-CoV S glycoprotein, isolated from transgenic mice expressing human immunoglobulin genes9,10. These transgenic mice contains human immunoglobulin genes and inactivated mouse heavy chain and kappa light chain genes (Bristol-Myers Squibb). Transgenic mice were immunized weekly with 10 mg of SARS-CoV spike protein and adjuvants for 6–8 weeks. Hybridomas were generated following a standard fusion protocol9. A panel of over 36 hybridomas were isolated based on various neutralization activities against SARS-CoV with lead antibodies showing protective potency in mice and hamster models9,10. To explore the possibility that some of the SARS-CoV-specific hybridoma may have cross-reactivity against SARS-CoV-2, these hybridomas were recovered and screened by ELISA against the SARS-CoV-2 spike protein. MAb362 was identified with cross-binding activity against both the RBD and S1 subunit of the SARS-CoV and SARS-CoV-2 spike proteins (Supplementary Table 1).
While both IgG and IgA are expressed at the mucosa, IgA is more effective on a molar basis and thus the natural choice for mucosal passive immunization as we recently demonstrated in other mucosal infectious disease20,21. To further characterize the functionality of MAb362, variable sequences of MAb362 were cloned into expression vectors as either IgG or monomeric IgA isotypes. Both MAb362 IgG and IgA were assessed in ELISA-binding assays against the RBD of the S1 subunit for SARS-CoV (S270–510) and SARS-CoV-2 (S319–541) (Fig. 1a, b). MAb362 IgA showed better binding activities, compared with its IgG counterpart against SARS-CoV-2 S319–541 (Fig. 1b). Assessment of the binding kinetics was consistent with the ELISA-binding trends. The binding affinity of IgA with RBD of SARS-CoV-2 is significantly higher (0.3 nM) than that of IgG (13 nM) due to a much slower dissociation rate as an IgA (Koff = 1.13 × 10−3 ± 1.06 × 10−4) compared with an IgG (Koff = 7.75 × 10−5 ± 5.46 × 10−5) (Fig. 1e, f). Of note, MAb362 IgA and IgG showed similar binding affinity with SARS-CoV S270–510 (Fig. 1c, d).
Fig. 1: Binding of MAb362 IgG and IgA to spikes of SARS-CoV and SARS-CoV-2.
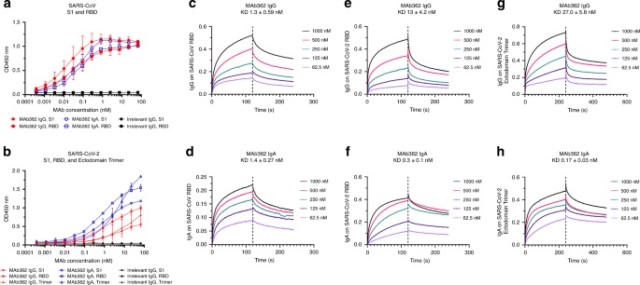
MAb362 IgG and IgA bind to purified SARS-CoV S1 (S1–590) and RBD (S270–510) truncations a and SARS-CoV-2 S1 (S1–604), RBD (S319–541), and ectodomain trimer b. IgGs are red lines, IgAs are blue lines, and irrelevant IgGs are black. Affinity measurements of MAb362 IgG c, e, g and IgA d, f, h against the RBD truncations of S glycoprotein of SARS-CoV and SARS-CoV-2 c–f, as well as ectodomain trimer of SARS-CoV-2 g, h were conducted using bio-layer interferometry and demonstrate nano and sub-nanomolar affinities. Data are plotted as the mean ± s.d. from n = 3 independent experiments a, b. Source data are provided as a Source Data file.
Full size image
To confirm binding results, the full ectodomain of spike was expressed including residues 1−1208 of SARS-CoV-2 with stabilizing proline mutations and a C-terminal T4 fibritin trimerization motif as described recently22 (Supplementary Fig. 1). MAb362 IgA still showed better binding activities with the stabilized trimer form as compared with its IgG isotype in ELISA (Fig. 1b) and affinity assays. The binding affinity of MAb362 IgA with the ectodomain of SARS-CoV-2 is 0.17 nM as compared with the 27 nM of IgG (Fig. 1g, h).
Structural modeling of MAb362 binding to RBD
To correlate the epitope binding with functionality, MAb362 IgG and IgA were tested in a receptor-blocking assay with Vero E6 cells. The result suggested that both MAb362 IgG and IgA block SARS-CoV-2 RBD binding to receptors in a concentration-dependent manner starting at ~30 nM (Fig. 2a, Supplementary Fig. 2a). Mutational scanning with a combination of alanine (to introduce a loss of interaction), tryptophan (to introduce a steric challenge), and lysine to introduce charge mutations were performed to better delineate the binding surface (Fig. 2b). The results showed that that key residues (Y449A, Y453A, F456A, A475W, Y489A, and Q493W) were critical for the complex and presumably, alterations in the packing caused marked loss of binding affinity (Fig. 2b and Supplementary Fig. 2b). Among the mutant we tested, A475W and Y489A also disrupted ACE2 binding (Supplementary Fig. 3). Interestingly, introduction of lysine mutations had little effect on binding, and some even showed enhanced binding, presumably owing to an overall more favorable charged interaction with the MAb362.

 - Neuntöter ♂")







")

